Regulatory Pathways to Promote Access to STI Diagnostics
Webinar Synopsis
There are an estimated 374 million new sexually transmitted infections (STIs) acquired per year. This equates to more than 1 million chlamydia, gonorrhea, syphilis, and trichomoniasis infections every day. These infections can lead to pelvic inflammatory disease, infertility, and increased transmission of HIV, when left untreated. The World Health Organization (WHO) publishes clinical guidelines to support STI testing for these infections, however, obtaining regulatory approvals for these diagnostics remains confusing. As the STI diagnostic landscape continues to evolve, it is imperative to review the processes that will help to promote access and uptake of new tests.

In September 2024, AVAC and WHO co-hosted a webinar to support researchers, product developers and the global advocacy community in identifying and discussing ways to bring new STI diagnostics to market with speed, equity and scale. Speakers have reviewed several pathways to create access to STI diagnostics, including:
- WHO prequalification;
- Expert Review Panel for Diagnostics; and
- Collaborative Registration Procedure.
Speakers’ experiences related to promoting access through regulations. The South African Health Products Regulatory Authority discussed ways to leverage these pathways and. The Bill and Melinda Gates Foundation highlighted the importance of building on these regulatory pathways and processes in catalyzing the development of new STI diagnostics and ensuring greater access to STI testing services.
Additional details are below and the recording of the webinar along with slides are available online.
WHO Prequalification of Diagnostics
- WHO prequalification (PQ) aims to promote and facilitate access to safe, appropriate, and affordable IVDs of good quality.
- The PQ application process includes several steps pictured below.
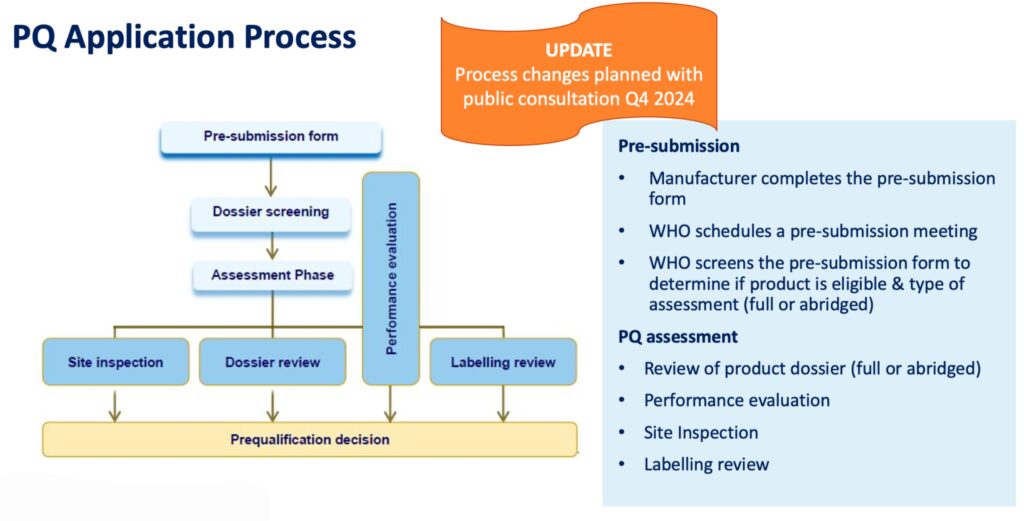
- In 2025, the WHO PQ application process will include STI diagnostics, both laboratory and point of care tests for chlamydia, gonorrhea, and trichomoniasis diagnosis using platforms (e.g. NAT; isothermal) for molecular detection and lateral flow and other technology for antigen testing.
- Technical Specification Series (TSS) documents are tailored to specific pathogen or type of assay and include requirements that address the needs of Member States in LMICs and related to general performance characteristics.
- The expert consultation to set the technical specifications for STI diagnostics was held in August 2024. The draft document will be available for public comment before the end of 2024. This is an important step which will advise developers on their PQ submissions for new products.

Expert Review Panel for Diagnostics
The purpose of the expert review panel for diagnostics (ERP-D) is to:
- Assess the potential risks/benefits associated with the procurement of diagnostic products that may have a high public health impact, but have not yet undergone a stringent assessment, either by the WHO Prequalification or by a SRA.
- Advise the Procurement Agencies (PA) and Technical Programs (TP) in their decision on whether to allow grant funds to be used for the time-limited procurement of the diagnostics reviewed by the ERPD.
- The ERPD risk/benefit assessment does not replace WHO PQ/SRA assessment but should be seen as a step towards a WHO PQ or full regulatory review.
- The ERPD mechanism should facilitate access to IVDs for neglected diseases, as well as innovative devices, if the associated risks are deemed to be lower than the potential benefits.
- Eligible products include innovative IVDs or IVD technologies with substantial public health impact that are listed through an Invitation to Manufacturer (ItM) for an Expression of Interest (EoI) that is developed and published by a PA or TP with IVD specifications.
Facilitated Registration Pathways and Collaborative Registration Procedure
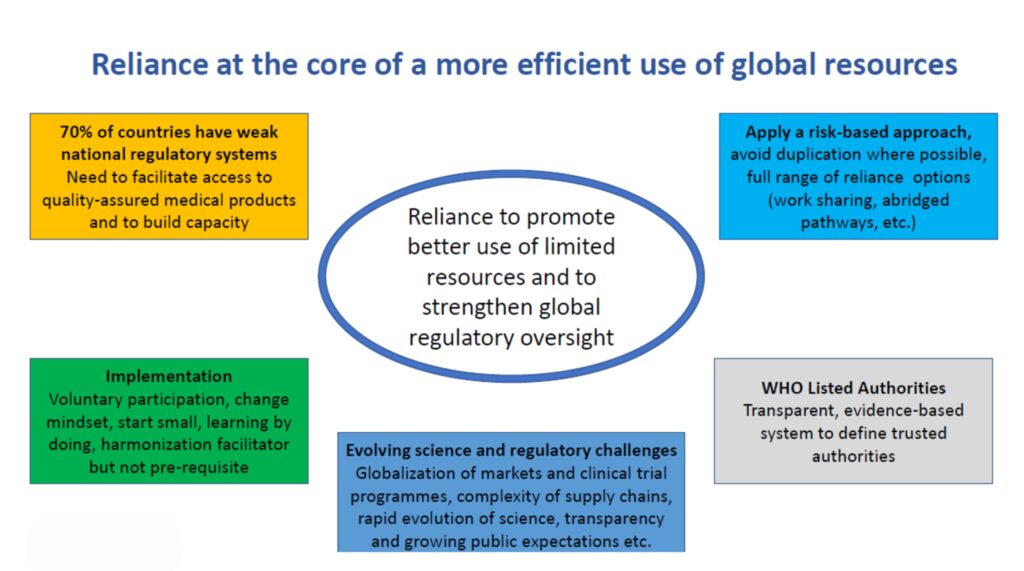
- Facilitated Regulatory Pathways (FRP) are a type of regulatory pathway available to National Regulatory Agencies (NRAs), which are meant to facilitate and accelerate the regulatory decisions and the introduction of quality-assured products in countries, through the use of concepts of reliance and collaboration.
- FRPs can help NRAs:
- Leverage on the work performed by others, improving efficiency of the regulatory systems by avoiding duplication of regulatory efforts and work.
- Optimize the use of human and financial resources and increase expertise and build capacities.
- Reduce the time needed to process a product application and reduce workload and backlog.
- Perform science-based and transparent regulatory decision-making, while maintaining national independence on their decisions.
- Ensure timely access to priority quality-assured products in countries.
- Collaborative Registration Procedure (CRP) facilitates the exchange of information to accelerate national registrations in countries through the provisions to NRAs of detailed assessment and inspection reports generated by reference NRAs/PQ
Perspectives from the South African Health Products Regulatory Authority (SAHPRA)
- LMICs are in frequent demand of prequalified products for the diagnosis of disease.
- More than 10 African NRAs participate in WHO PQ and all recognize WHO PQ for importation of IVDs into countries.
- Many of the regulatory systems need assistance and guidance to better regulate IVDs, facilitate access to quality assured medical products, and build capacity.
- Additionally, navigating some of the WHO processes can be confusing while balancing an evolving global regulatory space.
- WHO PQ has helped in strengthening regulatory systems, promoting the concept of reliance to increase effectiveness of regulation and efficiency, and informed NRAs own regulatory decision-making.
- WHO CRP has also led to shortening the approval time, i.e. the products are approved within 90 days from the day of submission in countries such Zambia, Tanzania, South Africa, and Kenya.
- Challenges around navigating the WHO PQ process include:
- Information asymmetry: Coordination and communication ,namely the lack of clarity in the procedure on some steps (who should notify whom), the lack of user-friendly tools to exchange information and report progress of applications, the applicants’ low level of awareness about the process and the lack of more direct interactions
- Quality of Submission: Quality issues that are occasionally discovered at the verification stage before being distributed. For example, manufacturers have been found to supply oral fluid HIV rapid diagnostic tests whose specifications were different from what was prequalified. The submissions of incomplete dossiers or discrepancies between the information submitted by the applicant and the one assessed by SRAs, were considered as the main causes for delays during the procedure. NRAs also identified the need for further guidance, particularly on post-approval management of IVDS approved through PQ.
- Double standards” – local vs. international market: Manufactures need to consider the needs of African markets along with other countries when developing products they hope to introduce into these countries.
- Post Approval Changes Management- Process not well understood by Manufacturers especially those in LMIC’s
- Lack of secure information technology systems: lack of secure platforms and procedures for the exchange and management of nonpublic information
- Lack of an assessment strategy and metrics for measuring and documenting the success of an agreement
- Resources misfit: Zambia only has two registration officers and can experience difficulties in adhering to timelines when receiving multiple submissions.
Recommendations
- Communicate about the added benefits-increased best practice sharing by WHO PQT
- Promote work-sharing initiatives among NRAs in LMICs (e.g., SADC/ZAZIBONA, ECOWAS, AMDF)
- Increase regular exchange of information and knowledge
- Strengthen formal procedure to collect input from NRAs on local unmet needs.
Resources
- Webinar Recording
- Webinar Slides: Introduction to Regulatory Pathways to Promote Accessibility of STI Diagnostics, Introduction to Facilitated Registration Pathways and Collaborative Registration Procedure (CRP) Slides, Prequalification of In Vitro Diagnostics Slides, Experiences from the South African Health Products Regulatory Authority, Expert Review Panel for Diagnostics (ERPD)
- Collaborative Procedure for Accelerated Registration: Includes information on the WHO collaborative procedure aims, principles, process, and impact.
- WHO collaborative registration procedure using stringent regulatory authorities’ medicine evaluation: reliance in action by Alexandra Vaz and colleagues: This article reports on the outcomes and stakeholders’ experience of using medicines assessments performed by Stringent Regulatory Authorities (SRA) in the Collaborative Registration Procedures (CRP).
FAQ
- Is there a full list of who the procurement agencies are who can make a request to ERPD? And can any technical program at WHO make a request to ERPD?
Answer: Request for ERPD assessment is typically made by agencies that are either WHO partners or those involved in global health procurement, such as Global Fund and GAVI, for diagnostics of interest for their needs. For new programmes or partners, WHO PQ team in charge of the ERPD program discuss the feasibility and funding source in consultation with procurement agencies and relevant WHO disease programmes. We also take into consideration our resources which are limited also.
- Is ERPD limited to tests for which the PQ team has already prepared a TSS?
Answer: ERPD is not limited to just IVDs that have a TSS in preparation and are in the PQ pipeline. ERPD has recently expanded to NTDs which may not necessarily enter the PQ pipeline. The decision of a round of ERPD and eligibility of a range of IVDs with substantial public health impact is determined in coordination with PQ team. But ultimately the Invitation for submitting an Expression of Interest is decided by the partner (procurement agency or WHO disease programme) who is the owner of the call.
- For TSS 24, will manufactures be able to submit for 1, 2 or 3 of the disease targets? I.e., can they submit for CT/NG as well as for CT/NG/TV, or must they include all 3?
Answer: For TSS 24 you can apply with tests that have any combination of the three disease targets, including just one.
- In what ways is STI regulatory different from other diagnostics?
Answer: The regulation of STI diagnostics does not differ to other IVDs. However, IVD regulations are changing in Europe which may have an impact on jurisdictions that rely on EU CE-Marking. Until recently manufacturers of STI diagnostics (chlamydia, gonorrhea and trichomoniasis) have been able to place their product in the market without having to submit their technical file for premarket approval in Europe. This is changing in Europe as new regulations are implemented. When diagnostics for STIs, including, chlamydia, gonorrhea, and trichomoniasis, become eligible for prequalification, they will undergo prequalification assessment applicable for a risk class C device (see our guidance on the prequalification assessment based on risk classification here)
- Is there a country list to facilitate registration through this procedure (CRP)?
Answer: List of countries that are currently participating in the CRP IVDS are found at Collaborative Procedure for Accelerated Registration | WHO – Prequalification of Medical Products (IVDs, Medicines, Vaccines and Immunization Devices, Vector Control). Additionally, other countries publish the WHO CRP process on their respective websites with processes to be followed, such as the Tanzania Medicines & Medical Devices Authority.
- What is the added value for manufacturers to go through WHO PQ (as opposed to regional bodies) considering timelines and cost?
Answer: WHO Prequalification aims to have a transparent review process, which clearly outlines the dossier, inspection, and laboratory evaluation requirements. If a product is prequalified, manufacturers also can leverage the collaborative registration procedure to facilitate registration in all interested countries. The list of countries who are participating is expanding with the aim of encompassing different regions. Prequalification assessment team is currently implementing efficiencies in the process, including recently applying recognition of MDSAP inspections.
- Which African countries do not require WHO PQ for public procurement? And if not, what regulatory authority do they recognize?
Answer: NRAs have several pathways to authorize use of IVDs in their countries, either though market authorization or special authorization. WHO PQ facilitates assessment of information during market authorization process and fastens regulatory decision. For many African countries including South Africa, Botswana, and Rwanda, approval from one of the stringent regulatory authorities is required. These regulatory authorities include the founding members of the International Medical Device Regulators Forum (IMDRF), such as the US Food and Drug Administration (United States), European Commission Directorate-General for Health and Food Safety (European Union), Therapeutic Goods Administration, Health Canada (Canada), and Pharmaceutical and Medical Devices Agency (Japan).
- Which STI diagnostics undergo the ERPD pathway?
Answer: Currently there is no invitation for expression of interest for chlamydia, gonorrhea or trichomoniasis IVDs. Through the Global Fund ERPD, EOIs are accepted for syphilis RDTs.
- What diagnostics are WHO pre-qualified?
Answer: The list of WHO prequalified IVDs, including diagnostics, can be downloaded from the WHO Prequalified In Vitro Diagnostics website.
- Is there a PQ process for STIs? Which ones?
Answer: IVD products currently eligible for submission for PQ include syphilis, hepatitis B, and human papillomavirus. In 2025, the WHO PQ assessment process plans to include STI diagnostics, both laboratory and point of care diagnostics to detection chlamydia, gonorrhea, and trichomoniasis nucleic acid and rapid diagnostic tests (lateral flow assays) for chlamydia or gonorrhea antigens.
Was this content helpful?
Tell us how we can improve the content.
Was this content helpful?
Thank you for your feedback!